家族性高コレステロール血症
Familial hypercholesterolaemia
2017年12月7日 Nature Reviews Disease Primers Article number: 17093 (2017) doi: 10.1038/nrdp.2017.93
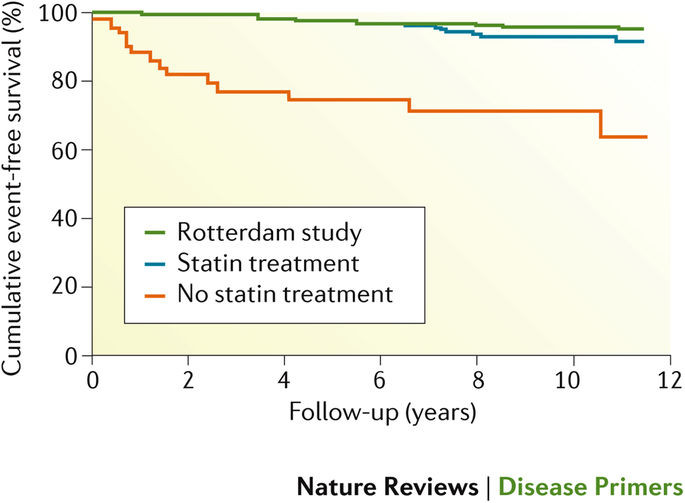
家族性高コレステロール血症はよくみられる遺伝性疾患の1つで、血中の低比重リポタンパク質(LDL)コレステロール値が生後から異常に上昇することを特徴とし、心血管疾患(CVD)の発症原因にもなる。大部分の症例でLDL受容体の遺伝子であるLDLRの常染色体優性変異が原因となっているが、コレステロール代謝やLDL受容体の機能とプロセッシングに関与するタンパク質の遺伝子(APOB 、PCSK9など)の変異も頻度は低いが原因とみられている。家族性高コレステロール血症の診断にはいくつかの基準がある。中でもLDLコレステロール値の上昇と高コレステロール血症または(若年性)CVDの家族歴がよく用いられる診断上の特徴である。原因となっている遺伝子異常を同定する遺伝子検査が望ましいが、診断法としては必須ではない。家族性高コレステロール血症患者の家族を対象とした早期診断にはカスケードスクリーニングが有用であり、数十年の無症状期間があることからも重要である。臨床的重症度は、他の要因よりも原因となる遺伝子変異の性質によって決まり、さらに変異の種類によっても影響を受ける。生涯にわたるLDLコレステロール低下療法はCVD無病生存率と同生存期間を大幅に改善する。第一選択薬はスタチンであるが、エゼチミブ、胆汁酸吸着剤、PCSK9阻害薬などの追加や他の新しい治療法も必要となることが多い。
PrimeView
このPrimeViewsでは、低比重(LDL)コレステロールの代謝に着目し、家族性高コレステロール血症の原因遺伝子変異によって、血中LDLコレステロールが十分に除去されなくなる機構について図説する。
本Primerの図解サマリー
Familial hypercholesterolaemia is a common inherited disorder characterized by abnormally elevated serum levels of low-density lipoprotein (LDL) cholesterol from birth, which in time can lead to cardiovascular disease (CVD). Most cases are caused by autosomal dominant mutations in LDLR, which encodes the LDL receptor, although mutations in other genes coding for proteins involved in cholesterol metabolism or LDLR function and processing, such as APOB and PCSK9, can also be causative, although less frequently. Several sets of diagnostic criteria for familial hypercholesterolaemia are available; common diagnostic features are an elevated LDL cholesterol level and a family history of hypercholesterolaemia or (premature) CVD. DNA-based methods to identify the underlying genetic defect are desirable but not essential for diagnosis. Cascade screening can contribute to early diagnosis of the disease in family members of an affected individual, which is crucial because familial hypercholesterolaemia can be asymptomatic for decades. Clinical severity depends on the nature of the gene that harbours the causative mutation, among other factors, and is further modulated by the type of mutation. Lifelong LDL cholesterol-lowering treatment substantially improves CVD-free survival and longevity. Statins are the first-line therapy, but additional drugs, such as ezetimibe, bile acid sequestrants, PCSK9 inhibitors and other emerging therapies, are often required.