骨形成不全症
Osteogenesis imperfecta
2017年8月18日 Nature Reviews Disease Primers Article number: 17052 (2017) doi: 10.1038/nrdp.2017.52
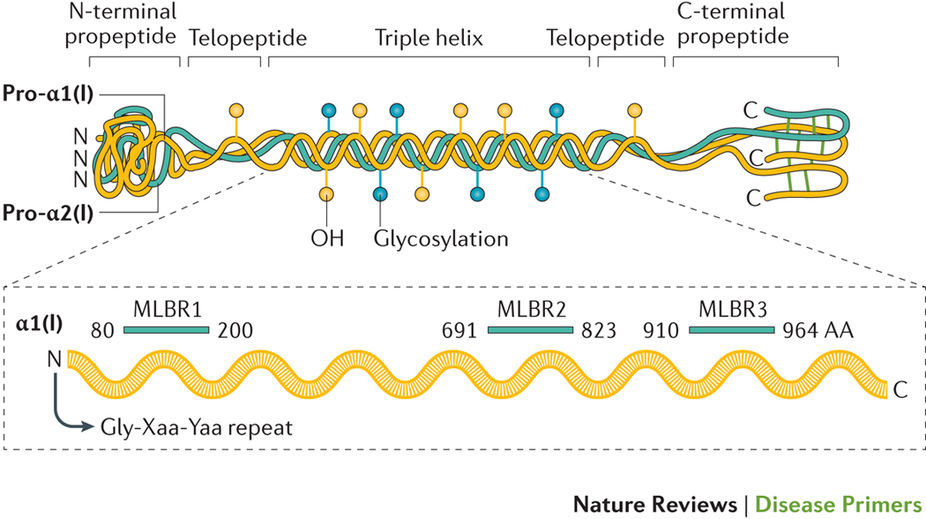
脆性骨疾患として知られる骨形成不全症は、骨の変形と脆弱化を特徴としている。骨形成不全症の診断は、通常、家族歴に加えて、出生前期、出生時または幼児期の骨折を特徴とする臨床像に基づいて行われる。遺伝学的検査によって診断を確定することもある。骨形成不全症患者の約85%は、コラーゲンの量または構造に影響を及ぼす、I型コラーゲン遺伝子(COL1A1およびCOL1A2)の常染色体優性突然変異が原因で発症する。この10年間に、I型コラーゲンの合成、プロセッシング、分泌および翻訳後修飾に関与するタンパク質遺伝子だけでなく、骨形成細胞の分化と働きを調節するタンパク質遺伝子を含む多種多様な遺伝子の劣性欠損(大部分がこれに相当する)や、優性およびX連鎖欠損が原因変異であることが報告されている。本疾患の原因遺伝子が多数存在することから、従来の疾患分類が困難になっており、新しい遺伝子分類システムが広く使用されてはいるが、現在も議論中である。骨の兆候だけでなく、心血管系や呼吸器系の異常、皮膚脆弱性、筋力低下、難聴および歯牙形成不全のように、さまざまな器官の表現型が報告されている。管理には、骨格異常に対する外科的および内科的治療に加えて、合併症の治療が行われる。現在、遺伝子治療や細胞療法をはじめ、シグナル伝達経路の変化に着目した、より革新的な治療法の開発が進められている。
PrimeView
脆性骨疾患として知られる骨形成不全症は、多様な遺伝子型と表現型を有する遺伝性骨疾患の一群である。このPrimeViewでは、この疾患の主な発症機構について解説する。
本Primerの図解サマリー
Skeletal deformity and bone fragility are the hallmarks of the brittle bone dysplasia osteogenesis imperfecta. The diagnosis of osteogenesis imperfecta usually depends on family history and clinical presentation characterized by a fracture (or fractures) during the prenatal period, at birth or in early childhood; genetic tests can confirm diagnosis. Osteogenesis imperfecta is caused by dominant autosomal mutations in the type I collagen coding genes (COL1A1 and COL1A2) in about 85% of individuals, affecting collagen quantity or structure. In the past decade, (mostly) recessive, dominant and X-linked defects in a wide variety of genes encoding proteins involved in type I collagen synthesis, processing, secretion and post-translational modification, as well as in proteins that regulate the differentiation and activity of bone-forming cells have been shown to cause osteogenesis imperfecta. The large number of causative genes has complicated the classic classification of the disease, and although a new genetic classification system is widely used, it is still debated. Phenotypic manifestations in many organs, in addition to bone, are reported, such as abnormalities in the cardiovascular and pulmonary systems, skin fragility, muscle weakness, hearing loss and dentinogenesis imperfecta. Management involves surgical and medical treatment of skeletal abnormalities, and treatment of other complications. More innovative approaches based on gene and cell therapy, and signalling pathway alterations, are under investigation.