結節性硬化症
Tuberous sclerosis complex
2016年5月26日 Nature Reviews Disease Primers Article number: 16035 (2016) doi: 10.1038/nrdp.2016.35
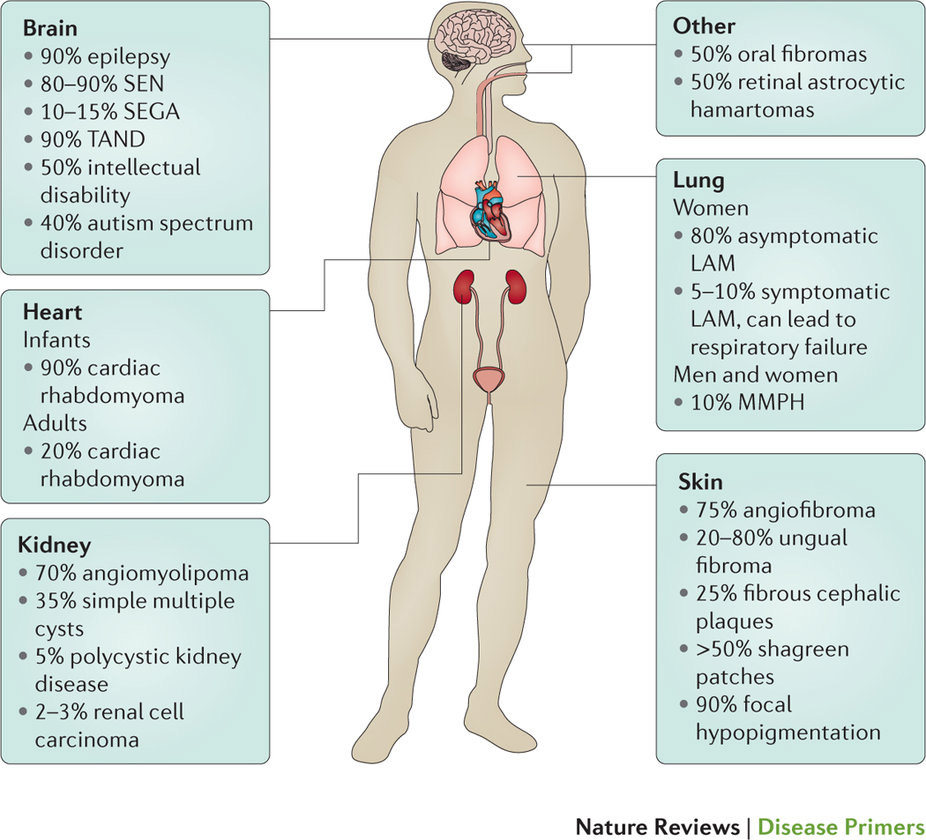
結節性硬化症(TSC)はさまざまな臓器系に影響を及ぼす常染色体優性遺伝疾患で、TSC1とTSC2のいずれか一方の遺伝子における機能喪失型変異が原因とされている。成人と小児のいずれもが罹患する疾患である。1880年にBournevilleが最初に報告して以来、現在では世界で約200万人の罹患者がいると推測されている。TSCの臨床像は特徴的であるが、たとえ同じ家族内であっても個々の患者により大きく異なっている。TSCの主徴には、脳、皮膚、心臓、肺および腎臓の腫瘍、てんかん、TSC関連神経精神障害(自閉症スペクトラム障害と認知障害を含める)などがある。TSC1(別名ハマルチン)およびTSC2(別名チュベリン)はTSC蛋白質複合体を形成してラパマイシン標的タンパク質(mTOR)のシグナル伝達経路を阻害することで、細胞成長、増殖、自己消化、および蛋白質と脂質の生成を調節する。基礎研究やトランスレーショナル研究の驚異的な進歩をはじめ、いくつかの多国間ランダム化対照試験によって、mTOR阻害薬は腎臓の血管筋脂肪腫、脳の星状細胞腫、および肺のリンパ脈管筋腫症の治療薬として承認されたが、すべての治療適応を取得するまでには、さらなる研究が必要である。このPrimerでは、疾患の分子的および細胞的基礎、医学的管理、主な知識格差および進行中の治療研究を含むTSC分野における最新の知識について取りまとめる。
PrimeView
このPrime Viewでは、TSC1とTSC2のいずれかの変異が原因となる生涯続く慢性疾患である結節性硬化症(TSC)の発症機構について取りまとめる。これらの変異はさまざまな臓器における良性腫瘍の増大を促進するシグナル伝達の変化につながっている。
本Primerの図解サマリー