網膜芽細胞腫
Retinoblastoma
2015年8月27日 Nature Reviews Disease Primers Article number: 15021 (2015) doi: 10.1038/nrdp.2015.21
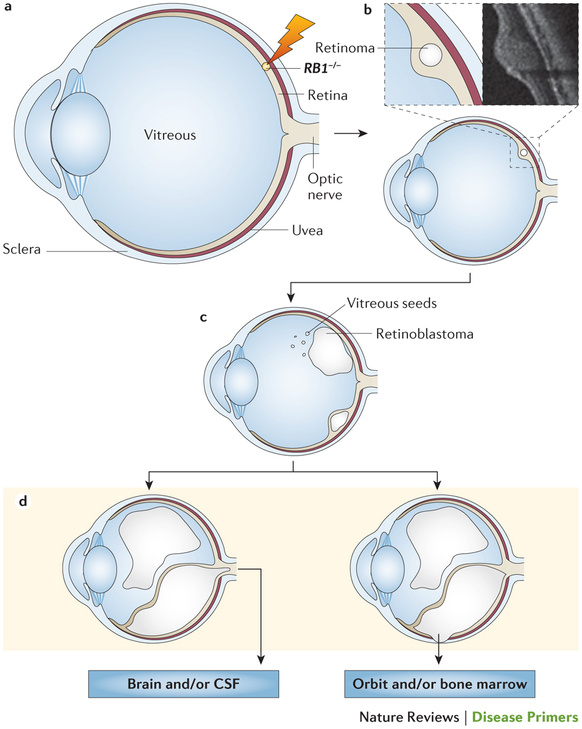
網膜芽細胞腫は、世界で毎年約8000人の子どもが診断される、小児の網膜に発生するまれに見られるがんの1つである。おそらく錐体前駆細胞のような形質転換感受性網膜細胞の網膜芽細胞腫遺伝子(RB1)の両アレルが変異すると網膜芽細胞腫が発生する。網膜芽細胞腫タンパク質(pRB)の腫瘍抑制機能が失われると、細胞分裂が制御不能になり、腫瘍の進行に伴ってゲノム変化が繰り返される。ほとんど全ての組織にpRBが発現しているが、錐体前駆細胞では、RB1喪失に対するpRBの感受性を高めて腫瘍形成を可能にするという生化学的および分子的性質が認められる。高所得国における患者の生存率は95%を超えるが、全世界では30%未満である。しかし、疾患啓発の向上による早期診断、新ガイドラインの活用、および臨床経験の共有によって、転帰は改善してきている。従来治療では失われていたかもしれない眼球を救出する有望な方法として、動脈内投与化学療法および硝子体内投与化学療法が登場した。現在進行中の国際的共同研究によって、眼病変の多種多様な分類は、治療法の適格性、有効性および安全性を一貫して評価するために標準化された分類定義に置き換えられるだろう。遺伝性網膜芽細胞腫の生存者は二次がんの発生リスクを有しているため、生涯にわたる経過観察が必要である。多様な組織におけるRB1喪失の分子レベルでの影響を明らかにすることによって、生殖細胞のRB1変異を有する患者の、二次がんに加えて、網膜芽細胞腫の治療と予防に新しい道が拓かれるだろう。
PrimeView
網膜芽細胞腫は、乳児または幼児に発生するまれな眼腫瘍で、網膜細胞におけるRB1の両アレルの喪失が主な原因である。このPrimeViewは、「網膜芽細胞腫」のPrimerを要約して、要点を明確にしている。
本Primerの図解サマリー