Review Abstract
嚢胞性線維症
Cystic fibrosis
2015年5月14日 Nature Reviews Disease Primers Article number: 15010 (2015) doi: 10.1038/nrdp.2015.10
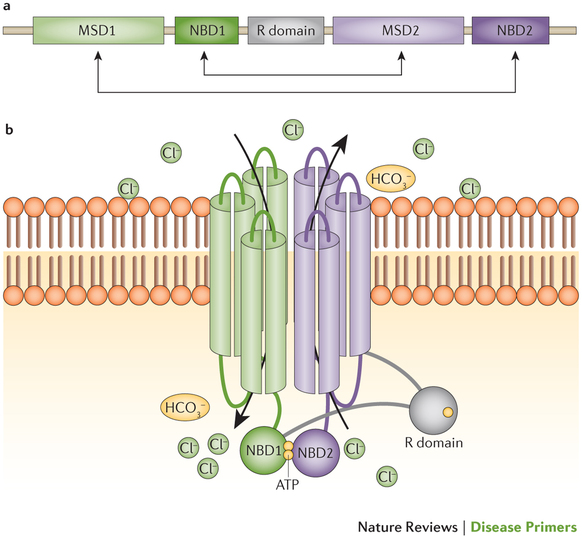
嚢胞性線維症は、常染色体劣性遺伝を示す単一遺伝子疾患で、嚢胞性線維症膜貫通調節因子(cystic fibrosis transmembrane conductance regulator )遺伝子(CFTR)の変異が原因で発症する。この遺伝子欠損が25年前に初めて報告されて以来、CFTR変異による発症機序や、その治療上の扱い方に関する私たちの理解は大きく進歩した。CFTRは膜貫通型タンパク質で、内皮細胞膜表面でイオンを輸送している。CFTR機能障害によって、さまざまな臓器が影響を受けるが、嚢胞性線維症患者の罹患と死亡の原因の圧倒的大部分は肺疾患によるものである。出生前診断、新生児スクリーニングおよび新しい治療アルゴリズムによって、本疾患の発症率と有病率は変わりつつある。嚢胞性線維症の標準ケアは、最近まで合併症の予防と治療に重点が置かれていた。しかし現在では、イオンチャネル異常を直接標的にした、新しい治療戦略が実施できるようになり、今後これらの治療法による疾患進行と患者のQOLへの影響を評価することが重要になると思われる。このPrimerでは、最新の知識を要約し、近い将来、新しい知識と治療選択肢が、嚢胞性線維症の臨床ケアと研究に及ぼす影響について、その展望を説明する。
Cystic fibrosis is an autosomal recessive, monogenetic disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. The gene defect was first described 25 years ago and much progress has been made since then in our understanding of how CFTR mutations cause disease and how this can be addressed therapeutically. CFTR is a transmembrane protein that transports ions across the surface of epithelial cells. CFTR dysfunction affects many organs; however, lung disease is responsible for the vast majority of morbidity and mortality in patients with cystic fibrosis. Prenatal diagnostics, newborn screening and new treatment algorithms are changing the incidence and the prevalence of the disease. Until recently, the standard of care in cystic fibrosis treatment focused on preventing and treating complications of the disease; now, novel treatment strategies directly targeting the ion channel abnormality are becoming available and it will be important to evaluate how these treatments affect disease progression and the quality of life of patients. In this Primer, we summarize the current knowledge, and provide an outlook on how cystic fibrosis clinical care and research will be affected by new knowledge and therapeutic options in the near future. For an illustrated summary of this Primer, visit: http://nature.asia/1LdF5zg