Research Abstract
次世代のシーケンシングは被覆度が低く、調節領域のSNP検出能力も低い
Next generation sequencing has lower sequence coverage and poorer SNP-detection capability in the regulatory regions
2011年8月5日 Scientific Reports 1 : 55 doi: 10.1038/srep00055
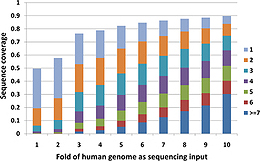
次世代シーケンシング(NGS)技術が急激に発展し、ゲノム研究の規模や精度が上昇する新たなチャンスが生まれている。NGSを最大限に活用するための2つの大きな課題は、読み取った何百万という短い塩基配列を参照ゲノム上にいかに効率よく貼り付けてマップを作るかと、どのように正確なSNPコーリングを行うかである。本論文では、マッピングとSNPコーリングのための現在のソフトウェアについて検討し、その性能をThe Cancer Genome Atlas(TCGA;がんゲノムアトラス)計画で得られた試料を用いて評価した。イルミナ社のNGSシステムに対する全般的な性能では、BWAとBowtieが、他のアラインメントツールよりも優れていることがわかった。一方、SOLiDに対する全般的性能はNovoalignCSが最も優れていた。また、次世代シーケンシングシステムは、ゲノムのCpGアイランド、プロモーター領域、5′-UTR領域において被覆度が著しく低く、SNPコーリングの性能も劣ることが明らかになった。これらの領域を対象にするNGS実験では、通常のゲノム領域よりも読みの深度を高くすべきである。
- 香港大学(中国)
- ニュージャージー工科大学(米国)
The rapid development of next generation sequencing (NGS) technology provides a new chance to extend the scale and resolution of genomic research. How to efficiently map millions of short reads to the reference genome and how to make accurate SNP calls are two major challenges in taking full advantage of NGS. In this article, we reviewed the current software tools for mapping and SNP calling, and evaluated their performance on samples from The Cancer Genome Atlas (TCGA) project. We found that BWA and Bowtie are better than the other alignment tools in comprehensive performance for Illumina platform, while NovoalignCS showed the best overall performance for SOLiD. Furthermore, we showed that next-generation sequencing platform has significantly lower coverage and poorer SNP-calling performance in the CpG islands, promoter and 5′-UTR regions of the genome. NGS experiments targeting for these regions should have higher sequencing depth than the normal genomic region.