神経芽腫
Neuroblastoma
2016年11月10日 Nature Reviews Disease Primers Article number: 16078 (2016) doi: 10.1038/nrdp.2016.78
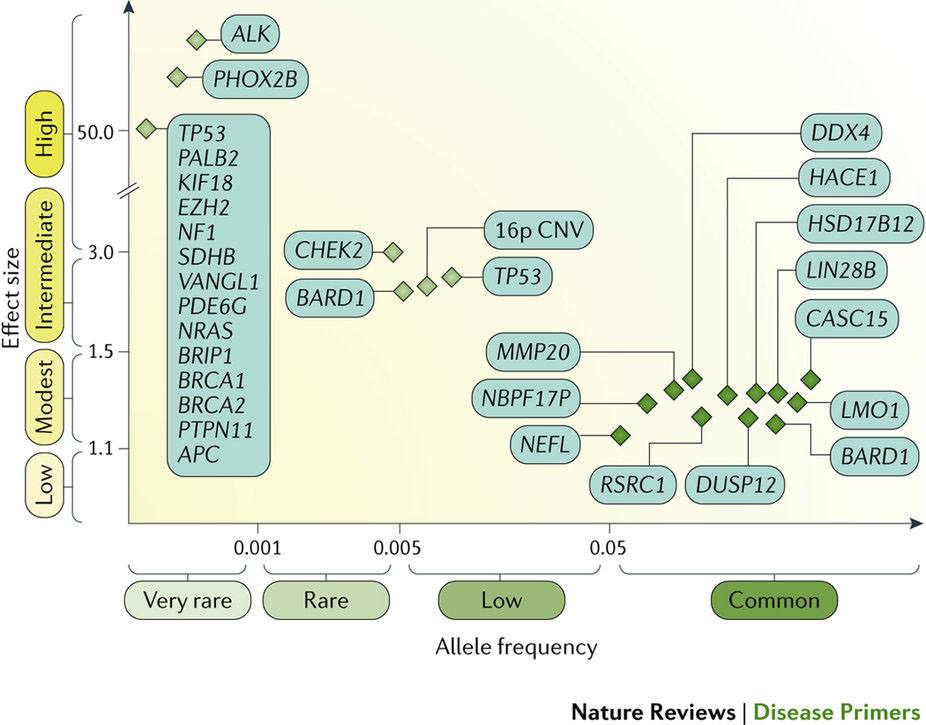
神経芽腫は小児期に最もよく見られる頭蓋外固形腫瘍であり、臨床像は多様で、腫瘍の生物学にしたがった経過をたどる。この神経内分泌腫瘍の特異な性質として、若年期に発症すること、診断時で既に転移していることが多いこと、および乳児では腫瘍が自然寛解する傾向があることが知られている。悪性度が最も高い腫瘍では転写因子をコードするMYCNがん遺伝子が増幅しており、通常、限局性腫瘍であっても予後不良と関連する。トランスジェニックマウスモデルではMYCNの過剰発現が腫瘍開始因子になるが、他の多くの関連遺伝子および腫瘍抑制遺伝子についても現在研究が進められており、これらの遺伝子も腫瘍発生に役割を担うとみられている。神経芽腫では染色体セグメントの異常が多く見られ、転帰不良との関連が示されている。家族性神経芽腫の発症はまれで、多くは ALKの生殖細胞変異と関連している。この遺伝子変異は原発腫瘍の10–15%に認められ、有望な治療標的になると考えられている。低リスクおよび中リスクの小児ではリスク層別化療法によって治療の軽減が促進されている。高リスク患者の治療も進歩しており、強力な化学療法や骨髄破壊的化学療法に続いて、分化誘導療法と免疫療法を使用した微小残存病変の治療が行われるようになった。これらの治療法よって5年全生存率は50%に改善している。現在、生存率と長期QOLのさらなる改善に向けて、ノルアドレナリン輸送体、遺伝経路および腫瘍の微小環境を標的とした新たな治療方法の開発に期待が集まっている。
PrimeView
神経芽腫は乳幼児期の神経内分泌腫瘍であり、副腎または交感神経節あるいはその両方に腫瘍が発生する。このPrimeViewでは、臨床検査、放射線イメージング技術および腫瘍生検などを含めた神経芽腫の診断法を中心に取りまとめる。
本Primerの図解サマリー
Neuroblastoma is the most common extracranial solid tumour occurring in childhood and has a diverse clinical presentation and course depending on the tumour biology. Unique features of these neuroendocrine tumours are the early age of onset, the high frequency of metastatic disease at diagnosis and the tendency for spontaneous regression of tumours in infancy. The most malignant tumours have amplification of the MYCN oncogene (encoding a transcription factor), which is usually associated with poor survival, even in localized disease. Although transgenic mouse models have shown that MYCN overexpression can be a tumour-initiating factor, many other cooperating genes and tumour suppressor genes are still under investigation and might also have a role in tumour development. Segmental chromosome alterations are frequent in neuroblastoma and are associated with worse outcome. The rare familial neuroblastomas are usually associated with germline mutations in ALK, which is mutated in 10–15% of primary tumours, and provides a potential therapeutic target. Risk-stratified therapy has facilitated the reduction of therapy for children with low-risk and intermediate-risk disease. Advances in therapy for patients with high-risk disease include intensive induction chemotherapy and myeloablative chemotherapy, followed by the treatment of minimal residual disease using differentiation therapy and immunotherapy; these have improved 5-year overall survival to 50%. Currently, new approaches targeting the noradrenaline transporter, genetic pathways and the tumour microenvironment hold promise for further improvements in survival and long-term quality of life.