血栓性血小板減少性紫斑病
Thrombotic thrombocytopenic purpura
2017年4月6日 Nature Reviews Disease Primers Article number: 17020 (2017) doi: 10.1038/nrdp.2017.20
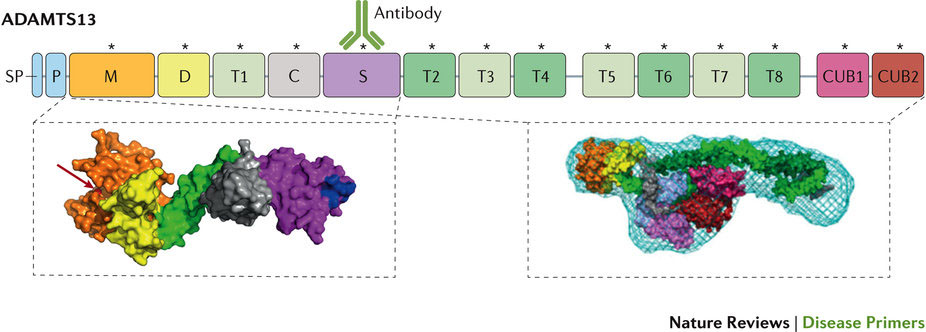
血栓性血小板減少性紫斑病(TTP、別名:Moschcowitz病)は、しばしば、重篤な血小板減少症や微小血管障害性溶血性貧血をはじめ、さまざまなレベルの虚血性臓器障害、とりわけ、脳、心臓、腎臓の障害が同時併発する特徴を有している。急性TTPは、患者のほとんどが死に至る疾患であったが、血漿療法が導入されてからは生存率が10%未満から80~90%にまで改善された。しかし、急性発作から回復した患者の再発リスクは高く、長期罹患率も高い。TTPでは、溶血性尿毒症症候群や他の血栓性微小血管症のような多くの疾患の症状と臨床所見も同時に発現するため、適時診断が極めて重要であるが困難である。TTPの病態生理は、von Willebrand因子(vWF)の多量体ストリングを切断するプロテアーゼであるトロンボスポンジンモチーフ13(ADAMTS13)を有するディスインテグリンおよびメタロプロテアーゼの重度の欠損である。内皮細胞から分泌されて接着した超大型vWFストリングは、切断されずに血小板と結合するため、微小血栓が形成されてTTPの臨床症状が発現される。先天性TTP(Upshaw-Schulman症候群)は、ADAMTS13のホモ接合性変異または化合物ヘテロ接合性変異によって引き起こされるが、後天性TTPは、酵素を阻害するか、またはその排泄を促進する血中の抗ADAMTS13自己抗体によって発症する自己免疫疾患である。そのため、後天性TTPでは血漿交換療法に加えてコルチコステロイドや、しばしばリツキシマブなどの免疫抑制薬が使用される。
PrimeView
血栓性血小板減少性紫斑病(TTP)は、まれに致死的となり得る微小血管症で、血小板減少症、溶血性貧血および虚血性臓器障害を特徴としている。このPrimeViewでは、特にTTPの確定診断の難しさについて解説する。
本Primerの図解サマリー
Thrombotic thrombocytopenic purpura (TTP; also known as Moschcowitz disease) is characterized by the concomitant occurrence of often severe thrombocytopenia, microangiopathic haemolytic anaemia and a variable degree of ischaemic organ damage, particularly affecting the brain, heart and kidneys. Acute TTP was almost universally fatal until the introduction of plasma therapy, which improved survival from <10% to 80–90%. However, patients who survive an acute episode are at high risk of relapse and of long-term morbidity. A timely diagnosis is vital but challenging, as TTP shares symptoms and clinical presentation with numerous conditions, including, for example, haemolytic uraemic syndrome and other thrombotic microangiopathies. The underlying pathophysiology is a severe deficiency of the activity of a disintegrin and metalloproteinase with thrombospondin motifs 13 (ADAMTS13), the protease that cleaves von Willebrand factor (vWF) multimeric strings. Ultra-large vWF strings remain uncleaved after endothelial cell secretion and anchorage, bind to platelets and form microthrombi, leading to the clinical manifestations of TTP. Congenital TTP (Upshaw–Schulman syndrome) is the result of homozygous or compound heterozygous mutations in ADAMTS13, whereas acquired TTP is an autoimmune disorder caused by circulating anti-ADAMTS13 autoantibodies, which inhibit the enzyme or increase its clearance. Consequently, immunosuppressive drugs, such as corticosteroids and often rituximab, supplement plasma exchange therapy in patients with acquired TTP.