22q11.2欠失症候群
22q11.2 deletion syndrome
2015年11月19日 Nature Reviews Disease Primers Article number: 15071 (2015) doi: 10.1038/nrdp.2015.71
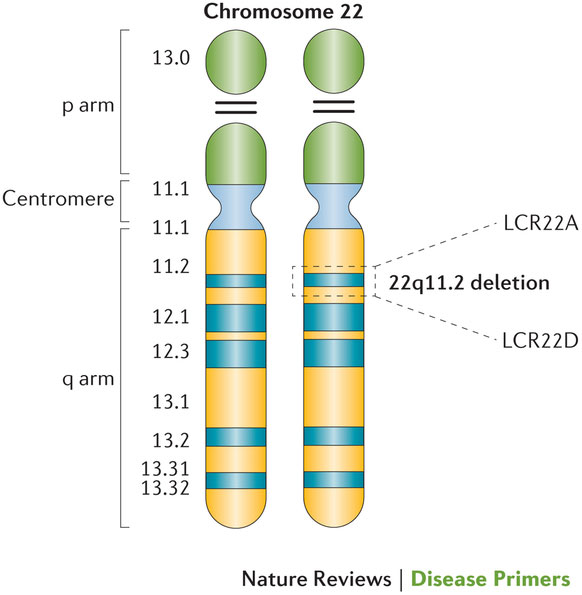
22q11.2欠失症候群(22q11.2DS)は、最もよく見られる染色体微細欠失疾患で、de novoの非相同性減数分裂期組換えイベントが主な原因と考えられており、胎児の1,000人に約1人の割合で発症する。現在ではこの染色体の違いが原因であると知られている一連の発見について、最初に英語で報告されたのは1960年代であり、それは、免疫不全症、副甲状腺機能低下症および先天性心疾患の臨床的三主徴を有するDiGeorge症候群の小児に関するものであった。この症候群には、他のさまざまな先天奇形および遅発性疾患(口蓋、消化器および腎臓の異常、自己免疫疾患、認知遅延、行動表現型および精神疾患など)の多様な症状が含まれることが示されており、いずれもDiGeorge症候群に関する最初の記述を拡大している。管理法には、小児科、総合内科、外科、精神科、心理学、介入療法(理学療法、作業療法、言語聴覚療法、行動療法など)および遺伝カウンセリングのような集学的アプローチが必要とされる。よくあることだが、この疾患に対する認識不足および/または遺伝子検査法への不慣れ、さらには臨床症状が多種多様であることが加わって、診断を遅らせている。早期診断、望ましくは出生前または新生児期の診断によって転帰の改善が見込めるため、全例スクリーニングの重要性が指摘されている。同様に重要なことは、22q11.2DSがさまざまな発症頻度の先天奇形、医学的症状、精神障害および発達障害を理解するモデルになっており、これらの障害を深く理解するためのプラットフォームを提供していることである。さらに、22q11.2DS患者をはじめ、これらの関連症状を有する一般集団の患者にも、生涯にわたってトランスレーショナル戦略の機会が与えられる。
PrimeView
22q11.2欠失症候群は臨床的に多様であり、とりわけ、口蓋異常、免疫機能不全、内分泌異常および認知遅延と行動遅延の原因になっている。このPrimeViewでは、22番染色体断片(22q11.2)の欠失に関与する病因論に焦点を合わせる。
本Primerの図解サマリー