The state of smart cities in MENA
11 December 2025
Published online 20 January 2020
A rare, fatal genetic disease is treated with an existing immunotherapeutic drug.
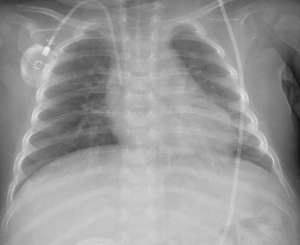
The New England Journal of Medicine ©2020
“We are in the renaissance period when it comes to rapid genetic diagnosis and experimental treatment of inherited disorders,” says associate professor of paediatrics Dusan Bogunovic of the Icahn School of Medicine at Mount Sinai, New York. “Now we know that USP18 deficiency is treatable with JAK inhibitors, if detected early.”
In healthy individuals, the USP18 (ubiquitin-specific peptidase 18) gene codes for an enzyme that inhibits a part of the immune system called interferon signalling, curbing excessive inflammation. USP18 enzyme deficiency leads to abnormal inflammation across multiple tissues. The Saudi child was diagnosed with complete absence of USP18, and had been kept alive through extraordinary medical care by physicians in Saudi Arabia. Genetic and biochemical tests confirmed that oral administration of ruxolitinib, a Janus kinase (JAK) inhibitor drug used for treating certain bone marrow disorders, would be suitable for regulating the child’s inflammation.
Within two months of ruxolitinib therapy, the boy was well enough to be weaned off of mechanical ventilation and is now, two years into therapy, in full remission of the clinical signs of the disorder. He is receiving follow-up treatment from an outpatient clinic and “continues to grow normally”, albeit “with slower progress in his developmental milestones,” the researchers say. The child will likely need to take ruxolitinib for the rest of his life.
Consultant paediatrician Abdullah Alangari at King Saud University, Saudi Arabia, says the study is “a very good example of the value of international collaboration in helping critically ill patients and in advancing science.”
Rare diseases cannot be managed alone, adds Bogunovic. “Each requires expertise that very few people on the planet have,” he says, commending the joint work by researchers in Saudi Arabia, France and the USA.
The clinical case study represents “a significant milestone,” says immunologist John Teijaro of Scripps Research Institute, USA, who was not involved in the study. “This success should pave the way for utilizing ruxolitinib and other JAK inhibitors to treat additional autoimmune diseases where heightened interferon or cytokine signalling drive pathological manifestations.”
doi:10.1038/nmiddleeast.2020.9
Alsohime, F. et al. JAK Inhibitor therapy in a child with inherited USP18 deficiency. New Engl. J. Med. 382, 256–265 (2020).
11 December 2025
11 December 2025
05 December 2025
21 December 2018
29 August 2018
22 June 2015
Sign-up to receive our e-alert update every two weeks to keep up with everything new on the portal.
Sign up for e-alerts
Stay connected: