鎌状赤血球症
Sickle cell disease
2018年3月15日 Nature Reviews Disease Primers 4 : 18010 doi: 10.1038/nrdp.2018.10
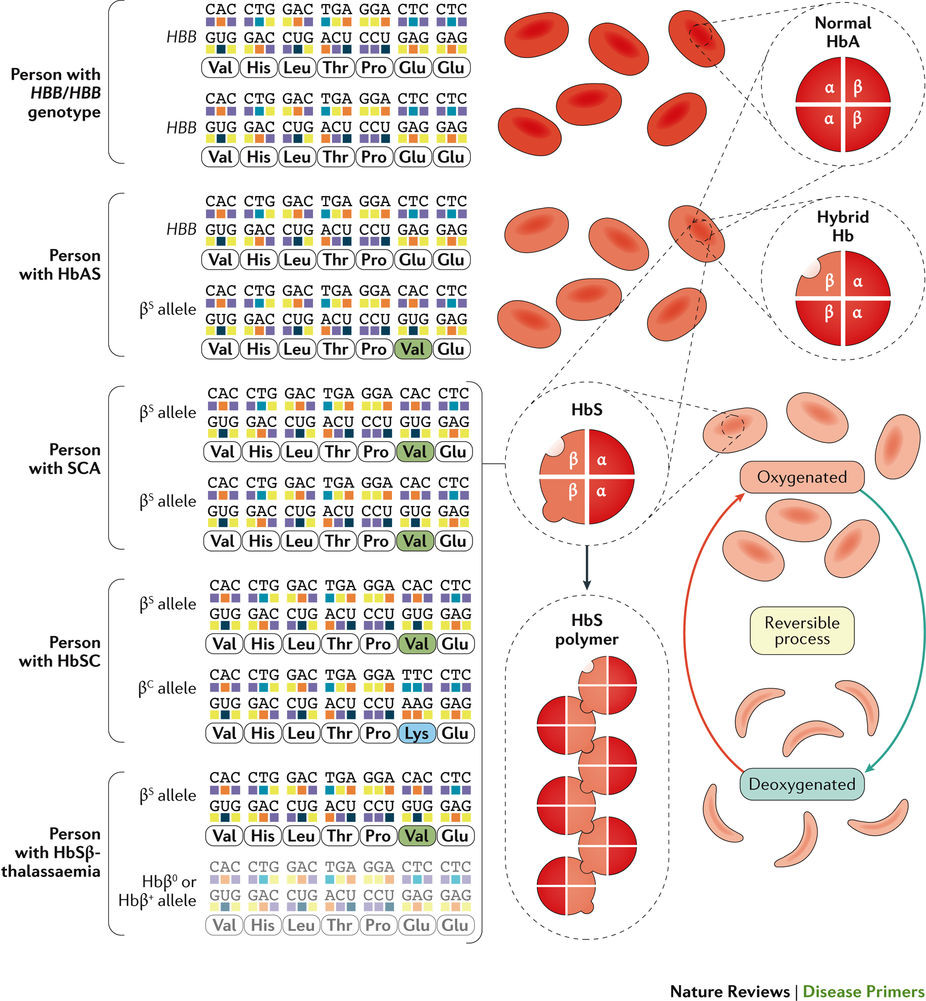
鎌状赤血球症は遺伝性疾患の1つで、ヘモグロビンのβサブユニット遺伝子である HBBの変異によって発症する。毎年世界で30万から40万例の新生児が発症しており、特にサハラ以南のアフリカに集中している。変異鎌状βグロビンサブユニットをもつヘモグロビン分子は重合化する性質を有している。内部のヘモグロビンの大部分が重合することで赤血球が鎌状になると考えられ、溶血が起こりやすくなる。これ以外にも、血管閉塞と免疫システムの活性化がSCDの表現型の病態生理学的機構に関与すると考えられている。このためSCDの表現型には非常に多くの合併症が現れる。よくみられる急性合併症は、疼痛発作、胸痛発作および脳卒中である。慢性腎臓病などの慢性合併症ではあらゆる臓器が障害される。ヒドロキシ尿素、輸血および造血幹細胞移植によってSCDの重症度が軽減することがある。生存率を改善するには早期の診断が重要であり、新生児の普遍的スクリーニングプログラムに取り組んでいる国もあるが、資金不足で医療資源が乏しい地域では実施が難しい。
PrimeView
このPrimeViewでは、ヘモグロビン変異体である鎌状ヘモグロビン(HbS)の重合化を原因とする鎌状赤血球症(SCD)の複雑な病態生理学的機構を中心に解説する。HbS重合体を内在する赤血球は鎌のような形になり、この疾患名の由来になっている。
本Primerの図解サマリー
Sickle cell disease (SCD) is a group of inherited disorders caused by mutations in HBB, which encodes haemoglobin subunit β. The incidence is estimated to be between 300,000 and 400,000 neonates globally each year, the majority in sub-Saharan Africa. Haemoglobin molecules that include mutant sickle β-globin subunits can polymerize; erythrocytes that contain mostly haemoglobin polymers assume a sickled form and are prone to haemolysis. Other pathophysiological mechanisms that contribute to the SCD phenotype are vaso-occlusion and activation of the immune system. SCD is characterized by a remarkable phenotypic complexity. Common acute complications are acute pain events, acute chest syndrome and stroke; chronic complications (including chronic kidney disease) can damage all organs. Hydroxycarbamide, blood transfusions and haematopoietic stem cell transplantation can reduce the severity of the disease. Early diagnosis is crucial to improve survival, and universal newborn screening programmes have been implemented in some countries but are challenging in low-income, high-burden settings.